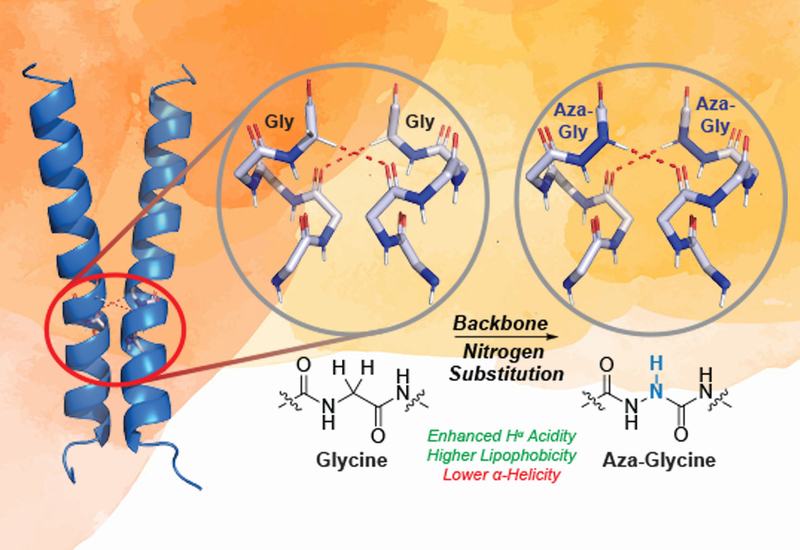
Transmembrane helix association governs the structure and activity of membrane proteins ranging from G-protein-coupled receptors to single-pass bitopic proteins. A recurring sequence feature at these interfaces is the GxxxG motif, in which two glycine residues separated by three residues orient toward the helix–helix contact surface. Because glycine lacks a side chain, it allows the two helices to approach closely, maximizing van der Waals packing, minimizing side-chain entropic cost, and exposing the backbone to the hydrophobic bilayer interior. That exposed backbone sustains a network of Cα–H···O=C hydrogen bonds between the two helices, and computational and isotope-edited infrared studies suggest these interactions contribute meaningfully to dimerization free energy. Yet experimental tools for perturbing GxxxG interfaces at the atomic level remain limited, and no reliable strategy exists for substituting the critical glycine residues, which have no mutable side chain to exploit.
Researchers in the Newberry Lab at The University of Texas at Austin, published in Biochemistry, reasoned that replacing the α-carbon with nitrogen to form aza-glycine would strengthen the hydrogen-bond donor at the dimer interface. Aza-glycine presents a more acidic Hα, analogous to the N–H donor of a conventional amide, and its greater polarity was expected to amplify solvophobic driving forces within the nonpolar bilayer. Precedent from collagen triple-helix studies, where aza-glycine substitution raised thermal stability by introducing new cross-strand hydrogen bonds, supported this reasoning. The team synthesized aza-glycine variants of two established model systems, glycophorin A, GpA, and Bcl-2 interacting protein 3, BNIP3, replacing Gly83 of GpA and Gly180 of BNIP3 with aza-glycine at the respective dimerization interfaces. Dimerization affinity was then measured via a Förster resonance energy transfer assay using pyrene and 7-dimethylaminocoumarin fluorophores attached to the peptide N termini.
Contrary to the design hypothesis, aza-glycine substitution reduced dimerization affinity in both systems. In SDS detergent micelles, the dissociation constant for aG-GpA was approximately 1.4-fold higher than for wild-type GpA, while the dissociation constant for aG-BNIP3 rose approximately 18-fold relative to wild-type BNIP3. Because the experimentally accessible detergent concentrations preclude fully sampling the monomeric and dimeric endpoints, the authors treat these values as semiquantitative comparisons rather than absolute affinities. To test whether the SDS micellar environment itself masked any stabilizing effects of aza-glycine, the team repeated the measurements in large unilamellar vesicles composed of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine. At three peptide/lipid ratios spanning a 100-fold range, aza-glycine-containing peptides showed consistently reduced Förster resonance energy transfer efficiency relative to their glycine counterparts for both GpA and BNIP3, confirming that the destabilizing effect is qualitatively independent of the membrane model system used.
Circular dichroism spectroscopy pointed toward the mechanistic explanation. Unlabeled aza-glycine peptides reconstituted in SDS micelles showed a 37% reduction in mean residue ellipticity at 222 nm for aG-GpA and a 31% reduction for aG-BNIP3 compared to their unmodified counterparts, indicating a loss of α-helical content. CD spectra recorded at 5 mM and 40 mM SDS, conditions that support different degrees of dimerization, were indistinguishable for both wild-type GpA and aG-GpA, demonstrating that the helicity decrease reflects intrinsic destabilization of the α-helix by the aza-glycine residue rather than merely a shift in the monomer–dimer equilibrium. Stereoelectronic effects of backbone nitrogen substitution bias the φ dihedral angle away from the α-helical region of conformational space, and this conformational cost appears to offset any enthalpic gain from enhanced hydrogen bonding. A competition Förster resonance energy transfer experiment, in which unlabeled peptides were titrated into labeled pairs to displace the dimer complex, corroborated these findings for GpA, though aG-BNIP3 displaced labeled wild-type BNIP3 to a similar extent as unlabeled wild-type BNIP3, suggesting greater tolerance for aza-glycine at the heterodimer interface relative to the homodimer.
The results clarify a tension that has persisted in the membrane protein folding field: stronger individual noncovalent contacts at a helix–helix interface do not automatically translate to tighter assembly if the modification simultaneously disrupts the secondary structure prerequisite for that interface. The findings also reveal that dimeric transmembrane helices tolerate aza-glycine substitution to a greater degree than β-sheet or β-turn scaffolds, implying that interhelical contacts provide compensatory stabilization. For the broader goal of engineering transmembrane peptidomimetics, the work establishes aza-glycine as a useful chemical probe for dissecting the relative contributions of backbone hydrogen bonding and helical propensity to GxxxG-mediated dimerization, even as it cautions that backbone nitrogen substitution alone is insufficient to enhance transmembrane helix assembly.